産品簡介
NF-κB由Ranjan Sen(NIH)在諾貝爾獎獲得者David Baltimore的實驗室中通過其與B細胞中免疫球蛋白輕鍊增強子中的II堿基對序列的相互作用而發現。
核因子-kB(NF-kB),是細胞内重要的核轉錄因子。它參與機體的炎症反應、免疫應答,能調節細胞凋亡、應激反應,NF-kB過度激活,與人類許多疾病如類風濕關節炎、心髒與腦部疾病的炎症變化等相關,因此通過藥物來抑制NF-kB信号轉導通路,可能會成為治療的手段。
NF-kB分子的N端含Rel同源域,參與其和DNA結合、參與二聚體化,能被NF-kB抑制物(TeB)結合、抑制;NF-kB分子内還有核輸出域、核定位域、轉位活性域等,C端有反式轉錄激活域。p50/p65NF-KB能與靶基因啟動子免疫球蛋白k輕鍊基因轉錄增強序列(kB序列)特異結合。RelA/c-Rel二聚體,能與靶基因啟動子其他序列結合。
NF-kB家族有5個成員,包括NF-kB1(p50)、NF-kB2(p52)、RelA(p65)、RelB和c-Rel,通常所說的NF-kB蛋白,是指p65/p50亞單位形成的NF-KB1二聚體蛋白;RelB/p52亞單位形成NF-kB2二聚體蛋白。
NF-kB可分兩組:p50/p52組,分别由pII0、p105前體裂解産生,p50/p52能與NF-kB家族其他成員形成二聚體,存留于胞質。RelA(p65),RelB和cRel一組,沒有前體。LkB是一種NF-kB的抑制蛋白,分子量36kD,可結合、抑制NF-kB并使NF-kB存留于胞質中,阻止NF-KB形成二聚體及入胞核,隻能在胞質組成p50-p65-1eBa/p複合體。高水平腫瘤壞死因子a、佛波酯、脂多糖、白介素2、H2O2,等,可激活NF-kB誘導性絲裂原蛋白激酶,再将IkBa/β磷酸化,磷酸化的IkBa/β的Lys殘基被泛素化後,可使IkBa/β降解,再使p50-p65NF-kB活化。
激活通路
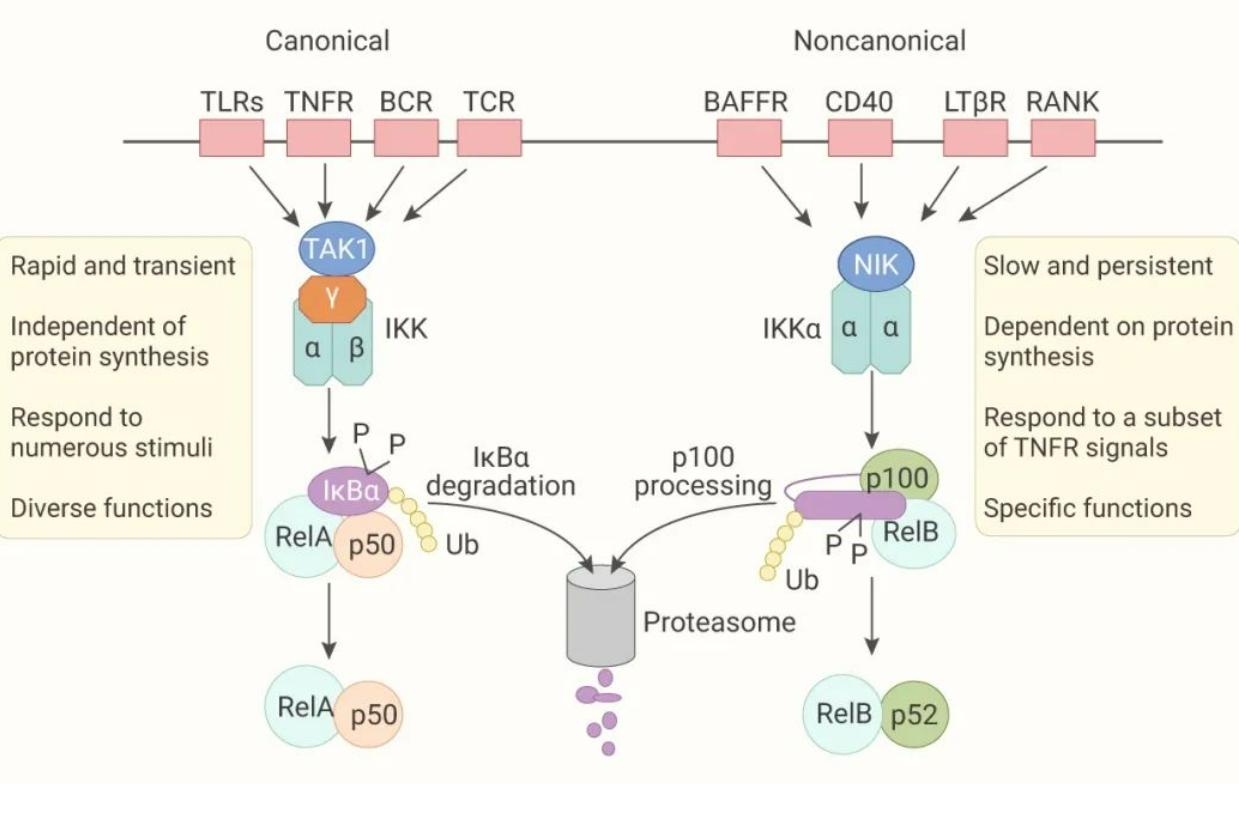
研究發現,激活NF-kB的信号轉導通路主要有以下三種:經典通路、旁路通路和非典型通路。NF-kB1,RelA,c-Rel,均由經典通路激活,而NF-kB2,RelB,由旁路通路激活,此外還有DNA氧化損傷等誘導的非典型通路;p65的翻譯後修飾,也調節NF-kB通路活性。
NF-kB信号通路激活的經典通路
當炎性因子腫瘤壞死因子a/白介素1/佛波酯/脂多糖等與相關受體結合後,引起後者構型改變,進而激活IkBa激酶,可使IkBa磷酸化,再在泛素連接酶P-TrCP的作用下泛素化,可被26S蛋白酶體識别并降解。于是NF-kB得以從細胞質NF-kB/IkBa複合物中釋放出來,并活化、暴露核定位域,形成p50/RelA二聚體,迅速發生核轉位,通過p50亞單位與靶基因的eB反應元件結合,從而啟動靶基因表達如腫瘤壞死因子a和白介素1等,5分鐘左右即可使細胞内NF-kB信号通路的活性水平達到峰值。蛋白激酶G/蛋白激酶C也可活化1kBa激酶、NF-kB
NF-kB信号通路激活的非經典通路
在腫瘤壞死因子受體TNFR家族配體,如CD40L,B細胞腫瘤壞死因子激活因子等的誘導下,相應的受體被激活,可促進NF-kB誘導的絲裂原蛋白激酶活化,再使蛋白激酶IKKa磷酸化,又使NF-KBp100被磷酸化降解,形成NF-eB的p52/RelB異源二聚體,又能使NF-eBp105被磷酸化降解,形成NF-kBp50/RelB異源二聚體,都進入細胞核從而調節靶基因的轉錄。CD40L,等還可活化NF-kB信号通路激活的經典通路。
NF-kB信号通路激活的其他通路
是指除了上述兩種通路以外的其他通路的總稱。适當水平的活性氧能活化酪氨酸激酶,再依次催化Rafl、蛋白激酶MAPKK/MAPK相繼磷酸化,使蛋白激酶p90S6被激活,蛋白激酶p90S6有蛋白激酶IKK樣活性,可使kBa的Tyr2殘基磷酸化,結果1kBa被降解,NF-KB得以釋放,繼而發生核轉位,與DNA上的kB反應元件結合,調節靶基因表達。該信号通路隻在少數敏感性細胞中存在。
血管緊張素II可通過AT,R/活性氧通路及酪氨酸激酶Src/蛋白激酶P13K通路,激活NF-kB,上調白介素6、細胞間黏附分子-1等的表達,可促炎症、促進血管平滑肌細胞增殖。腫瘤壞死因子受體可通過腫瘤壞死因子受體相關因子(TRAF2/5/6)激活NF-KB,可促炎症,抗凋亡。Toll樣受體信号通路活化、DNA損傷、UV輻射、蛋白激酶P13K、表皮生長因子受體(EGFR)、蛋白激酶C、内質網應激等,也可激活NF-wB通路。蛋白激酶A、蛋白激酶C,MSK1也可影響NF-cB活化的程度。p300/帽子結合蛋白CBP,p300/帽子結合蛋白CBP相關因子等,可使活化的NF-KBp50乙酰化,可增加NF-kBp50與DNA的結合力。NF-kB作用的靶基因有200多種,大緻可分為以下六類,主要參與免疫應答、炎症反應、細胞增殖、抗凋亡、血管發生、腫瘤侵襲和轉移等。rB反應元件存在于靶基因啟動子中。
阻斷策略、應用
通過抑制IKK而抑制NF-KB的活性
将IkBa激酶-IKK的顯性負突變體導入細胞中,可特異性阻斷NF-cB的活化;針對NEMO(1KKy)的抗灸多肽,可與NEMO結合,從而阻止NEMO與IKKp的連接,可抑制急性炎症反應。小分子ATP競争性抑制物,可抑制IKKp活性,進而抑制NF-kB活性,這為炎症反應的治療提供了新方法。
通過kBa抑制NF-kB活性
這方面研究熱點,是給予IkBa突變體、IcBa超級抑制劑,抑制NF-kB活性。
通過抑制蛋白酶體的活性抑制NF-KB活性
磷酸化的IkBa可被泛素蛋白酶體降解。泛素蛋白酶體抑制劑二肽硼酸類、廣譜泛素蛋白酶體抑制劑MG-132,可抑制磷酸化的IkBa可被泛素蛋白酶體降解,能抑制NF-kB的活性。
通過對NF-kB的調控抑制NF-kB活性
對NF-KB的調控主要通過三個方面,即抑制其磷酸化、阻斷其核定位及與DNA的結合、抑制靶基因表達。免疫抑制劑PG490,是從中藥雷公藤中提取的一種二帖環氧化物,能選擇性地作用于NFB的p65亞單位,抑制p65的轉錄激活域,在NF-KB與DNA結合之後,雷公藤可抑制靶基因的轉錄。一些藥物如芍藥苷,可抑制NF-kB的表達,從而可減輕由慢性腦血流灌注不足引起的腦損傷。合成與靶基因的順式調控NF-KB元件相似的寡脫氧核糖核苷酸,将其轉導入細胞,能在NF-kB入核前後,與NF-kB競争,阻斷NF-KB與靶基因啟動子的結合活性,從而抑制靶基因的轉錄,可減輕炎症,已成功用于抑制炎症性休克,治療再灌注損傷、心腦梗死等。
其他抑制NF-kB活性的策略
應用抗氧化劑,如N-乙酰多巴胺二聚體,可以抑制免疫和炎症反應中NF-kB的活性。探索具有靶細胞特異性和對不同NF-KB成員具有選擇性的細胞内NF-kB活化阻斷劑,可為臨床治療開辟新的途徑。
研究發現,NF-kB調節的靶蛋白包括:
1.促凋亡因子,如Bax、胱冬蛋白酶II、CD95、死亡受體Fas、死亡配體FasL,GADD45g、癌蛋白-Myc,p5、腫瘤壞死因子受體1,TRAF結合蛋白TRAIL。
2.抑凋亡因子,如Bel-2,Bcl-xL,Bfl,c-FLIP、凋亡誘導蛋白IAPs,白介素13、腫瘤壞死因子a、腫瘤壞死因子受體1、腫瘤壞死因子受體相關因子TRAF1/2/6,IEX
3.細胞周期控制因子如癌蛋白cMyc,Rel、幹擾索調控因子IRF-4,p21CIP1、周期索DI、周期素D2、周期素D3、ephrin-A1,GADD45β
4.生長因子,如白介素1/2/6/8/9/II/12/15、白介素2受體、正常T細胞活化下調蛋白RANTEs、粒細胞-巨噬細胞集落刺激因子。
5.黏附分子,如細胞間黏附分子(ICAM-1)/血管細胞間黏附分子(ICAM-V)/E-選擇素。
6.促轉移因子,如尿激酶型纖溶酶原激活物(u-PA)
7.促血管生長因子,如血管内皮生長因子(VEGF)
研究發現,NF-kB的誘導的受體包括:B細胞受體、T細胞受體、Toll樣受體1~II、核苷酸結合域蛋白(NOD1)、腫瘤壞死因子受體1/2/4-1BB,Baff-R,CD27,CD30,CD40,死亡受體Fas/DR4/R5,外異蛋白A受體(EDAR),XEDAR,LTPR,OX40、白介素1受體、核因子受體活化蛋白受體RANK,RELT型腫瘤壞死因子受體、TIR、白介素18受體、Thelper1(THI)、補體C3a受體、補體C5a受體、KSHV-G蛋白耦聯受體、表皮生長因子受體、整合素a5p1/a53/a6p4,p2整合素、趨化因子受體CXCR1/CXCR2/CXCR6、緩激肽受體(B2)、神經生長因子受體(p75,TrkA)、粒細胞一巨噬細胞集落刺激因子受體、血小闆源性生長因子受體、溶血磷脂酸受體等。
臨床意義
癌症
NF-κB被真核細胞廣泛用作控制細胞增殖和細胞存活的基因調節因子。因此,許多不同類型的人類腫瘤具有錯誤調節的NF-κB:即,NF-κB具有組成型活性。活性NF-κB啟動基因的表達,使細胞保持增殖并保護細胞免受通過細胞凋亡導緻其死亡的條件。在癌症中,控制NF-κB信号傳導的蛋白質發生突變或異常表達,導緻惡性細胞與其他生物體之間的協調缺陷。這在轉移以及免疫系統對腫瘤的低效根除中都是明顯的。當正常細胞從它們所屬的組織中移除時,或當它們的基因組不能與組織功能協調地運作時,它們會死亡:這些事件依賴于NF-κB的反饋調節,并且在癌症中失敗。
NF-κB的缺陷導緻細胞凋亡的易感性增加,導緻細胞死亡增加。這是因為NF-κB調節抗凋亡基因,特别是TRAF1和TRAF2,因此消除了caspase酶家族的活性,這是大多數細胞凋亡過程的核心。
在腫瘤細胞中,NF-κB具有活性(例如,41%的鼻咽癌),這是由于編碼NF-κB轉錄因子本身的基因突變或控制NF-κB活性的基因(如IκB)基因);此外,一些腫瘤細胞分泌導緻NF-κB活躍的因子。阻斷NF-κB可導緻腫瘤細胞停止增殖,死亡或對抗腫瘤劑的作用更敏感。因此,作為抗癌治療的靶标,NF-κB是制藥公司中許多活躍研究的主題。
然而,盡管令人信服的實驗數據已經确定NF-κB是腫瘤發生的關鍵啟動子,這為基于抑制NF-κB活性的抗腫瘤治療的發展創造了堅實的理論基礎,但在考慮抗NF時應謹慎行事。 -κB活性作為癌症治療中的廣泛治療策略,因為數據還顯示NF-κB活性增強腫瘤細胞對凋亡和衰老的敏感性。此外,已經顯示經典NF-κB是Fas轉錄激活因子,而替代NF-κB是Fas轉錄抑制因子。因此,NF-κB促進癌細胞Fas介導的細胞凋亡,并因此抑制NF-κB的抑制可以Fas介導的細胞凋亡損害宿主免疫細胞介導的腫瘤抑制。
炎症
因為NF-κB控制着很多與炎症有關的基因,所以發現NF-κB在許多炎症性疾病中具有慢性活性也就不足為奇了,例如炎症性腸病,關節炎,敗血症,胃炎,哮喘,動脈粥樣硬化等。 。值得注意的是,一些NF-κB活化劑(如骨保護素(OPG))的升高與死亡率升高有關,尤其是心血管疾病。升高的NF-κB也與精神分裂症有關。最近,NF-κB活化被認為是香煙煙霧在骨骼肌中分解代謝作用的可能分子機制。肌肉減少症。研究表明炎症期間的細胞的功能取決于它的信号響應激活與相鄰細胞和激素的組合,特别是通過特定的受體作用于細胞因子它接觸。組織内的細胞表型通過反饋信号的相互刺激而發展,反饋信号與其他細胞協調其功能;當組織暴露于炎症時,這在細胞功能的重編程期間尤其明顯,因為細胞改變它們的表型,并逐漸表達在消除炎症原因後準備組織再生的基因組合。特别重要的是在組織駐留細胞和免疫系統的循環細胞之間發展的反饋反應。不同細胞類型和免疫系統之間反饋反應的保真度取決于限制NF-κB激活的基因範圍的機制的完整性,隻允許表達有助于有效免疫反應的基因,并随後完成炎症消退後恢複組織功能。在癌症中,調節基因表達響應炎症刺激的機制被改變到細胞停止将其存活與協調其表型及其功能的機制與組織的其餘部分聯系起來的程度。這在NF-κB活性的嚴重受損調節中通常是明顯的,其允許癌細胞表達NF-κB靶基因的異常群組。這不僅導緻癌細胞異常運作:周圍組織的細胞改變其功能并且僅停止支持生物體。另外,癌症微環境中的幾種類型的細胞可以改變它們的表型以支持癌症生長。因此,炎症是一種測試組織成分保真度的過程,因為導緻組織再生的過程需要協調不同細胞類型之間的基因表達。
NEMO
NEMO缺陷綜合征是一種罕見的遺傳病,與IKBKG中的缺陷有關,後者又激活NF-kB。它主要影響男性,并具有高度可變的症狀和預後。
成瘾
NF-κB是ΔFosB的幾種誘導的轉錄靶标之一,其促進發展和維持對刺激的成瘾。在尾殼核中,NF-κB誘導與運動增加有關,而在伏隔核中,NF-κB誘導通過獎勵緻敏增強藥物的積極增強作用。