簡述
聚合酶鍊式反應(PCR)是20世紀80年代後期由K.Mullis等建立的一種體外酶促擴增特異DNA片段的技術。PCR是利用針對目的基因所設計的一對特異寡核苷酸引物,以目的基因為模闆進行的DNA體外合成反應。由于反應循環可進行一定次數,所以在短時間内即可擴增獲得大量目的基因。PCR技術具有靈敏度高、特異性性強、操作簡便等特點。雖然PCR技術也存在出錯傾向高、産物大小受到限制和必須預先有目标DNA序列等缺點,但仍被譽為20世紀分子生物學研究領域最重大的發明之一。Mullis也因貢獻卓著而獲得1993年度諾貝爾獎。目前,PCR技術已廣泛應用于分子生物學的各個領域,在分子克隆、法醫學鑒定、DNA序列分析、緻病基因的檢測及考古學等方面都發揮着重要作用。
發展曆程
Khorana(1971)等最早提出核酸體外擴增的設想:“經DNA變性,與合适的引物雜交,用DNA聚合酶延伸引物,并不斷重複該過程便可合成tRNA基因。”n但由于當時基因序列分析方法尚未成熟,熱穩定DNA聚合酶尚未報道以及引物合成的困難,這種想法似乎沒有實際意義。加上分子克隆技術的出現提供了一種克隆和擴增基因的途徑,所以Khorana的設想被人們遺忘了。n1983年4月的一個星期五晚上,他開車去鄉下别墅的路上,猛然閃現出“多聚酶鍊式反應”的想法。n1983年12月,Mullis用同位素标記法看到了10個循環後的49bp長度的第一個PCR片段。n1985年,KaryMullis在Cetus公司工作期間,發明了PCR。Mullis要合成DNA引物來進行測序工作,卻常為沒有足夠多的模闆DNA而煩惱。n1985年10月25日申請了PCR的專利,1987年7月28日批準(專利号4,683,202),Mullis是第一發明人。n1985年12月20日在Science雜志上發表了第一篇PCR的學術論文,Mullis是共同作者。n1986年5月,Mullis在冷泉港實驗室做專題報告,全世界從此開始學習PCR的方法。
步驟
DNA的半保留複制是生物進化和傳代的重要途徑。在生物體内,雙鍊DNA在多種酶的作用下可以變性解鍊成單鍊,在DNA聚合酶與啟動子的參與下,根據堿基互補配對原則複制成同樣的兩分子挎貝。在實驗中發現,DNA在高溫時也可以發生變性解鍊,當溫度降低後又可以複性成為雙鍊。因此,在生物體外,通過溫度變化控制DNA的變性和複性,并設計引物做啟動子,加入DNA聚合酶、dNTP就可以實現特定基因的體外複制。
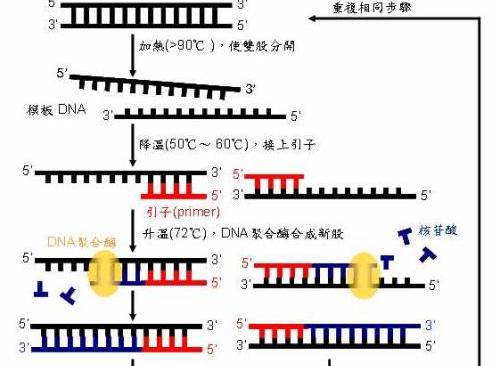
PCR由變性--退火--延伸三個基本反應步驟構成:n①模闆DNA的變性:模闆DNA經加熱至94℃左右一定時間後,使模闆DNA雙鍊或經PCR擴增形成的雙鍊DNA解離,使之成為單鍊,以便它與引物結合,為下輪反應做準備;n②模闆DNA與引物的退火(複性):模闆DNA經加熱變性成單鍊後,溫度降至55℃左右,引物與模闆DNA單鍊的互補序列配對結合;n③引物的延伸:DNA模闆--引物結合物在Taq酶的作用下,以dNTP為反應原料,靶序列為模闆,按堿基配對與半保留複制原理,合成一條新的與模闆DNA鍊互補的半保留複制鍊。n重複循環變性--退火--延伸三過程,就可獲得更多的“半保留複制鍊”,而且這種新鍊又可成為下次循環的模闆。每完成一個循環需2~4分鐘,2~3小時就能将待擴目的基因擴增放大幾百萬倍。
反應物質
反應體系:10×擴增緩沖液10ul、4種dNTP混合物各200umol/L、引物各10~100pmol、模闆DNA0.1~2ug、TaqDNA聚合酶2.5u、Mg2+1.5mmol/L、加雙或三蒸水至100ul
參加PCR反應的物質主要有五種即引物、酶、dNTP、模闆和緩沖液(其中需要Mg2+)
引物
PCR産物的特異性取決于寡核苷酸引物與模闆DNA互補的程度。引物設計的基本原則是最大限度地提高擴增效率和特異性,同時盡可能抑制非特異性擴增。反應中的引物至少應含有18個與模闆序列完全互補的核苷酸(常用軟件通常默認為20個),最大不能多于38個,否則最适延伸溫度會超過TaqDNA聚合酶的最适溫度(74℃),不能保證PCR擴增産物的特異性這樣才能保證擴增反應的特異性。引物擴增跨度以500核苷酸為宜,特定條件下可擴增長至10kb的片段。引物堿基的G+C含量以40-60%為宜,太少擴增效果不佳,過多易出現非特異條帶。同時ATGC最好随機分布,避免5個以上的嘌呤或嘧啶核苷酸的成串排列,尤其3′端不應超過3個連續的G或C,因為這樣會使引物在G+C富集區引發錯誤延伸。控制引物的濃度,避免引物内部出現二級結構和引物間互補。引物3’端的堿基要求嚴格配對(不能做任何修飾),引物5′端可修飾,加上限制内切酶位點、啟動子序列或其它序列等,以便于PCR産物的分析克隆。
DNA聚合酶
最常用的是耐熱TaqDNA聚合酶。目前有兩種市面上有兩種TaqDNA聚合酶供應,一種是從水生嗜熱杆菌中提純的天然酶;另一種是由大腸菌合/成的基因工程酶。一個典型的PCR反應約需酶量1-2.5U/100ul體系,濃度過高可引起非特異性擴增,濃度過低則合成産物量減少。Taq酶在95℃持續溫育仍能保持活性,因此引物的退火和延伸可以在更高的溫度下進行,使引物與模闆的誤配大大減少。然而TaqDNA酶5’→3’聚合酶活性和5’→3’外切酶活性,無3’→5’外切活性,在PCR反應中如發生某些堿基的錯配,該酶是沒有校正功能的。保真性不如PfuDNA聚合酶等。
此外還有VentDNA聚合酶和PfuDNA聚合酶。
四種dNTP
在PCR反應中,dNTP應為50~200μM,濃度過低會降低PCR産物的産量。配制過程注意4種dNTP的濃度要相等(等摩爾配制),如其中任何一種濃度不同于其它幾種時(偏高或偏低),就會引起錯配。而高濃度的dNTP可與Mg2+結合,使遊離的Mg2+濃度下降,影響DNA聚合酶的活性。
模闆DNA
模闆DNA的來源有三種,可從微生物中或血細胞、絨毛、尿樣、毛發、精斑、口腔上皮細胞等細胞中提取DNA,也可将固定和包埋的組織标本脫蠟、蛋白酶K消化後提取DNA。模闆DNA的濃度常為0.1~2ug/100ul體系。PCR産量随模闆DNA濃度的增加而顯著升高。
Mg2+
Mg2+對PCR擴增的特異性和産量有顯著的影響。在一般的PCR反應中,各種dNTP濃度為200umol/L時,Mg2+濃度為1.5~2.0mmol/L為宜,Mg2+濃度過高,反應特異性降低,出現非特異擴增,濃度過低會降低TaqDNA聚合酶的活性,使反應産物減少。
影響因素
溫度與時間
标準反應中采用三溫度點法,雙鍊DNA在90~95℃變性,再迅速冷卻至40~60℃退火,然後快速升溫至70~75℃延伸,對于較短靶基因(長度100~300bp)可采用二溫度點法,将退火與延伸溫度合二為一,一般采用94℃變性,65℃左右退火與延伸。變性溫度與時間一般93℃~94℃,1min足以使模闆變性。若低于93℃則需延長時間,但溫度不能過高,因為高溫環境對酶的活性有影響。退火溫度是影響PCR特異性的重要因素。退火(複性)溫度與時間取決于引物的長度、堿基組成及其濃度,還有靶基因序列的長度。對于20個核苷酸,G+C含量約50%的引物,55℃作為選擇最适退火溫度的起點較為理想。一般試驗中退火溫度比擴增引物的融解溫度TTm低5℃,可按公式進行計算:Ta=Tm-5℃=4(G+C)2(A+T)-5℃
其中A,T,G,C分别表示相應堿基的個數。例如,20個堿基的引物,如果(GC)%含量為50%時,則Ta的起點可設在55℃。在典型的引物濃度時(如0.2μmol/L),退火反應數秒即可完成,長時間退火沒有必要,一般為30~60s,足以使引物與模闆之間完全結合。延伸溫度一般選擇在70~75℃之間,延伸溫度高于90℃時,DNA合成幾乎不能進行。過高的延伸溫度不利于引物和模闆的結合。延伸反應時間根據待擴增片段長度而定,1Kb以内的DNA片段,延伸時間1min(足夠),3~4kb的靶序列需3~4min,擴增10kb需延伸至15min。延伸時間過長會導緻非特異性擴增。對低濃度模闆的擴增,延伸時間要稍長些。
循環次數
PCR反應的循環次數主要取決于模闆DNA的濃度,一般為25~35次,此時PCR産物的積累即可達到最大值。
增強劑
PCR反應中加入一定濃度的增強劑如DMSO(1%~10%)、甘油(5%~20%)、非離子去污劑、甲酰胺(1.25%~10%)和牛血清白蛋白(10~100μg/ml)等可提高反應特異性和産量,有些反應隻能在這些輔助劑存在時才能進行。但需要注意的是,這些增強劑濃度過高時不僅不能提高PCR反應的特異性和産量,還會對PCR反應産生抑制作用。
熱啟動PCR
即首先将模闆變性,然後在較高溫度時加入TaqDNA聚合酶、引物及MgCl2等一些重要成分,這樣使得引物在較高溫度下與模闆退火,提高了反應的嚴謹性,使擴增更特異。
應用模式
兼并引物PCR(Degenerate Primer PCR)
密碼子具有兼并性,單以氨基酸順序推測編碼的DNA序列是不精确的,但可以設計成對兼并引物,擴增所有編碼已知順序的核酸序列。用兼并引物時寡核苷酸中核苷酸序列可以改變,但核苷酸的數量應相同。兼并度越低,産物特異性越強,設計引物時應盡量選擇兼并性小的氨基酸,并避免引物3’末端兼并,針對兼并的混合引物已成功地用于未知靶DNA的擴增、克隆和序列分析。現已成功地克隆了豬尿酸氧化酶基因、糖尿病相關肽基因和哺乳動物與禽類的嗜肝病毒基因。用脫氧肌苷(deoxyinosine;DI)引物進行PCR,可以代替編碼蛋白的多種兼并密碼子中的兼并堿基,DI的特異性主要受cDNA濃度影響。
套式引物(Nested Primer PCR)
用第一套引物擴增15~30個循環,再用擴增DNA片段内設定的第二套引物擴增15~30個循環,這樣可使待擴增序列得到高效擴增,而次級結構卻很少擴增。用起始引物限量方法或Centricon30(Amicon)分子濾過器離心,在第二套引物加入前去除第一引物。此方法已成功地用來分析中國倉鼠卵巢細胞AS52的分子突變。AS52細胞含有單拷貝的細胞gpt(guaninephos-phribosytransferase)基因,與哺乳動物具有同源性。套式引物PCR減少了引物非特異性退火,從而增加了特異性擴增,提高了擴增效率。對環境樣品中微生物檢測和單拷貝的基因靶DNA的擴增是非常有效的。
若将套式PCR的内外引物稍加改變,延長外引物長度(至25~30bp),同進縮短内引物長度(15~17bp),使外引物先在高溫退火溫度下做雙溫循環擴增,然後改換至三溫循環,使内引物在外引物擴增的基礎上作低溫火溫度的三溫循環直到擴增完成,這樣就可以使兩套引物一次同時加入,兩種循環一氣呵成,等于隻做一次PCR,而靈敏度與套式二次PCR無異,在我們最近推出的PTc51氣流式DNA熱循環儀上就可以完成全部程序。套式一次PCR的成功,使PCR檢測的全過程可以在5h内完成,使當天出檢驗報告成為現實,也使PCR檢測走入臨床有了現實的基礎。
複合PCR(Multiplex PCR)
用多對引物同時擴增幾條DNA片段的方法稱為複合PCR。這一方法最初是由Chanberlain等檢測人的基因發展而來。Bej等随之發展了對環境樣品中不同屬細菌相關基因序列同時PCR擴增的檢測方法。兩種不同的軍團菌(legionella)基因,一為特異嗜肺L基因(mip),另一種為L-5SrRNA基因,通過引物搖擺(staggered)添加進行複合PCR。首先mip引物PCR擴增7個循環,然後加入5SrRNA引物PCR擴增38個循環。加入不同量的LacZ和LacB基因引物進行PCR擴增可以檢測大腸杆菌和與人類糞便污染有關的細菌包括E.coli大腸菌、腸源緻病沙門氏菌和志賀氏菌。
在複合PCR中,所有引物Ta值應相近。如果兩對引物Tq值差異超過±℃10%,會使擴增産物的量明顯不同,其中一種擴增産物或目的DNA很難觀察到。另外,靶DNA的長度也應相近,差别大時短片的靶DNA會優先擴增,因此,會産生不同産量的擴增産物,為此,須采用DNA搖擺擴增或加入不等量的引物方法進行解決。
反向PCR(Inverse PCR或Reverse PCR)
反向PCR的目的在于擴增一段已知序列旁側的DNA,也就是說這一反應體系不是在一對引物之間而是在引物外側合成DNA。反向PCR可用于研究與已知DNA區段相連接的未知染色體序列,因此又可稱為染色體緩移或染色體步移。這時選擇的引物雖然與核心DNA區兩末端序列互補,但兩引物3’端是相互反向的。擴增前先用限制性内切酶酶切樣品DNA,然後用DNA連接酶連接成一個環狀DNA分子,通過反向PCR擴增引物的上遊片段和下遊片段;現已制備了酵母人工染色體(YAC)大的線狀DNA片段的雜交探針,這對于轉座子插入序列的确定和基因庫染色體上DNA片段序列的識别十分重要。
該方法的不足是:①需要從許多酶中選擇限制酶,或者說必須選擇一種合适的酶進行酶切才能得到合理大小的DNA片段。這種選擇不能在非酶切位點切斷靶DNA。②大多數有核基因組含有大量中度和高度重複序列,而在YAC或Cosmid中的未知功能序列中有時也會有這些序列,這樣,通過反向PCR得到的探針就有可能與多個基因序列雜交。
利用反向PCR可對未知序列擴增後進行分析,探索鄰接已知DNA片段的序列,并可将僅知部分序列的全長cDNA進行分子克隆,建立全長的DNA探針。适用于基因遊走、轉位因子和已知序列DNA旁側病毒整合位點分析等研究。
不對稱PCR(Asymmetric PCR)
不對稱PCR的基本原理是采用不等量的一對引物産生大量的單鍊DNA(ss-DNA)。這兩種引物分别稱為限制性引物與非限制性引物;其最佳比例一般為1:50~1:100,關鍵是限制引物的絕對量。限制性引物太多太少,均不利于制備ss-DNA。也可用普通PCR制備靶DNA雙鍊DNA(ds-DNA),再以ds-DNA為模闆,隻用其中一種過量引物進行單引物PCR制備ss-DNA。産生的ds-DNA與ss-DNA由于分子量不同可以在電泳中分開,而得到純ss-DNA。不對稱PCR主要為測序制備ss-DNA,尤為用cD-NA經不對稱PCR進行DNA序列分析是研究真核DNA外顯子的好方法。
标記PCR(LP-PCR)和彩色PCR
LP-PCR(Labelled Primers PCR)是利用同位素、熒光素等對PCR引物5’端進行标記,據此檢測目的基因的存在與否,與常規PCR相比更為直觀,省去了限制性内切酶酶切及分子雜交等繁瑣步驟,而且一次可以同時分析多種基因成分,因而特别适合于大量臨床标本的基因診斷。該方法隻對PCR産物進行定性鑒定。
彩色PCR(Color complementassay)直譯為“着色互補性檢測”,是LP-PCR的一種,彩色PCR意譯更為明确:它用熒光染料标記引物的5’端。熒光染料JOE和FAM呈綠色熒光;TAMRA呈紅色熒光;COUM呈藍色熒光。不同熒光标記的引物同時參加反應,擴增後的目的基因會分别帶有引物5’端的染料,通過電泳或離心沉澱,肉眼就可以根據不同熒光的色澤判斷目标基因是否存在及擴增基因的類型。通常僅需2種不同顔色的引物,一種作為基因檢測引物;另一種作為控制條件的内對照,即可診斷基因缺失、染色體易位或感染某種病毒。檢測多種點突變時,可用更多的色彩,如多點突變的遺傳病、幾種可疑病毒感染、HLA位點分析都可以用彩色PCR同時檢測多個位點。
加端PCR
加端PCR(add-PCR)是使擴增産物的5’-末端加一段DNA順序的PCR。設計加端PCR的引物時,除與模闆配對的那一部分外再加上若幹堿基,這樣使擴增産物的末端加上額外一段DNA,如加上一個限制酶的識别順序或特定功能的DNA片段。Stoflet等報道在結構基因前加上噬菌體T7的啟動子,當然也可用于DNA片段的末端标記或引入特定的點突變。末端可加堿基的數量與引物的長短有關,當引物足夠長時擴增産物或末端甚至可以加上十幾個到幾十個堿基。
錨定PCR或固定PCR
錨定PCR(Anchored PCR,A-PCR)主要用于分析具有可變末端的DNA序列,Loh等用A-PCR對人外周血淋巴細胞T細胞受體α-鍊的mRNA的多變性進行了分析。先合成cDNA,并用末端脫氧核苷酸轉移酶在其3’-可變區末端加上一個PolyG尾巴。Loh等恒定區與可變區連接部位設一個引物,另一個引物是一個具5’-polyG尾巴的引物。帶有PolyG尾巴的引物是一個固定點,它可以并與PolyG尾巴結合,無論其餘部分序列如何,隻識别片段末端,利用此法可從前述mRNA中檢出至少20種不同序列,每一種都是獨特的,表明A-PCR不對任何特殊序列有傾向性結果,可用于T細胞、腫瘤及其它部位抗體基因的研究。
玻片PCR
在聚丙烯管中可以對多種含膜闆材料進行PCR,而在顯微玻片上用組織細胞塗片或切片直接進行DNA擴增的方法就稱為玻片PCR(Slide-PCR)。先将細胞塗片或呈單層細胞,然後用甲醇/醋酸(3:1,V/V)、Carnoy溶液、無水乙醇或4%多聚甲醛溶液固定5~15min。用蒸餾水沖洗,幹燥,直接使用或保存于-20℃備用。在玻片上劃20mm×28mm為免疫組化反應區。加入30μlPCR反應混合液,其中含10mmol/LTrispH8,3,50mmol/LKCl,1.5mmol/LMgCl2,200μmol/ldNTPs,100nmol/L引物,0.01%(V/V)明膠,0.2%BSA,2.5u/100μlTaq酶。然後蓋上22mm×40mm的蓋玻片,邊緣用石蠟油封好。把玻片放入PCR熱循環儀金屬塊上,使金屬塊與樣品呈最大程度接觸,同在聚丙烯管中一樣,進行30~40個循環。對于較短的擴增片段在後期循環中變性溫度可降低。反應後,将緻冷玻片放在氯仿中除去大部分石蠟油,但不取出蓋玻片,用一個尖鑷子輕輕拈起蓋玻片一角,在相對的一角中PCR反應混合液呈半月形液面,用移液器回收。一般可回收25μl混合液,将反應産物進行瓊脂糖電泳或用套式PCR引物按标準PCR進行重新擴增。片上擴增物可做原位雜交顯示。
在Slide-PCR中,需0.1%~1%的BSA。加入BSA可以保證擴增結果,但效率不一定很高。明膠(至少0.0001%),對擴增1kb左右的靶DNA十分重要。但對小片段擴增結果影響不大。不同的樣品提取方法或固定法對Slide-PCR都可行。
Silde-PCR的機理可能是在起始變性過程中一部分DNA從細胞中洗提出來,然後在細胞和玻片的水相中進行PCR。用地高辛标記的人全基因組DNA探針雜交表明在起始循環中DNA極微量,而30個循環後很豐富。常規細胞染色表明隻有少量的形态改變。
Silde-PCR對于玻片上的細胞樣品提供了一種較好的方法,而不必再把這些樣品從玻片上括下來,使操作簡便,污染減少。本方法對于原樣品量極微且需病史追蹤保存的(如子宮頸塗片或塗片)具有實用價值。
反轉錄PCR方法檢測RNA
RNA的多聚酶鍊式反應(RT-PCR)是以RNA為模闆,聯合逆轉錄反應(reversetranscrip-tion,RT)與PCR,可用于檢測單個細胞或少數細胞中少于10個拷貝的特異DNA,為RNA病毒檢測提供了方便;并為獲得與擴增特定的RNA互補的cDNA提供了一條極為有利和有效的途徑。RNA擴增包括兩個步驟:①在單引物的介導下和逆轉錄酶的催化下,合成RNA的互補鍊cDNA;②加熱後cDNA與RNA鍊解離,然後與另一引物退火,并由DNA聚合酶催化引物延伸生成雙鍊靶DNA,最後擴增靶DNA。
在RT-PCR中關鍵步驟是RNA的逆轉錄,cDNA的PCR與一般PCR條件一樣。由于引物的高度選擇性,細胞總RNA無需進行分級分離,即可直接用于RNA的PCR。但RT-PCR對RNA制品的要求極為嚴格,作為模闆的RNA分子必須是完整的,并且不含DNA、蛋白質和其它雜質。RNA中即使含有極微量的DNA,經擴增後也會出現非特異性擴增;蛋白質未除淨,與RNA結合後會影響逆轉錄和PCR;殘存的RNase極易将膜闆RNA降解掉。硫氰酸胍(GaSCN)-CsCl法或酸性硫氰酸胍-酚-氯仿法可提得理想的RNA制品,尤以後者方法為佳,适合一般實驗室進行。
常用的逆轉錄酶有兩種,即禽類成髓細胞性白血病病毒(Avianmyeloblastosisvirus,AMV)和莫洛尼鼠類白血病病毒(Moloneymurineleukemiavirus,MO-MLV)的逆轉錄酶(RT)。一般情況下用Mo-MLV-RT較多,但模闆RNA的二級結構嚴重影響逆轉錄時,可改用AMV-RT,因後者最适溫度為72℃,高于Mo-MLV-RT的最适溫度(37℃),而較高的反應溫度有助于消除RNA的二級結構。
一步法擴增(onestepamplification)是為了檢測低豐度mRNA的表達,利用同一種緩沖液,在同一體系中加入逆轉錄酶、引物、Taq酶、4種dNTP直接進行mRNA反轉錄與PCR擴增。發現Taq酶不僅具有DNA多聚酶的作用,而且具有反轉錄酶活性,可利用其雙重作用在同一體系中直接以mRNA為模闆進行反轉錄和其後的PCR擴增,從而使mRNA的PCR步驟更為簡化,所需樣品量減少到最低限度,臨床小樣品的檢測非常有利。用一步法擴增可檢測出總RNA中小于1ng的低豐度mRNA。該法還可用于低豐度mRNA的cDNA文庫的構建及特異cD-NA的克隆,并有可能與Taq酶的測序技術相組合,使得自動反轉錄、基因擴增與基因轉錄産物的測序在一個試管中進行。
定量PCR
1、DNA-PCR定量用同位素标記的探針與電泳分離後的PCR擴增産物進行雜交,根據放射自顯影後底片曝光強弱可以對模闆DNA進行定量。Abbot等利用這種方法對人類T細胞白血病反轉錄基因進行了定量研究。
PCR擴增産物用專為檢測ds-DNA而設計的微量熒光計定量,利用染料H33258專與雙鍊DNA結合而使熒光增強50倍的特性。可以從标準模闆系列稀釋擴增産物量曲線上讀出樣品中模闆DNA的量或拷貝數,達到PCR定量的目的。
利用倍比稀釋模闆作系列稀釋PCR,求出最低(PCR-EB)檢測限來比較,也是常用的半定量PCR方法。
2、mRNA-PCR定量由于MRNA-PCR定量需經兩個酶(RT和Taq)催化,因而影響因素較多。1989年Wang等報道了低豐度mRNA絕對定量方法。利用濃度已知且與待測靶mR-NA序列相同的内對照mRNA(其片段長短不同,便于PCR擴增後産物的分離),在同一體系中,用相同的由32P标記的引物與待測mRNA一同進行逆轉錄和PCR擴增,擴增産物電泳後,分别測定二者産物放射性強度,由預先制備的标準曲線推算出每個樣本特異mRNA的量。Gilliland等的結果表明,在1ng總RNA中可以對小于1pg的特異mRNA進行定量。這一定量方法在腫瘤、代謝失調、基因表達調控等研究中均有重要意義。
技術應用
由于PCR技術具有快速、簡便、靈敏等特點,已被廣泛地應用于臨床醫學、遺傳咨詢、司法鑒定、考古學及分子生物學等各個領域,有的替代了原有的方法,有的大大簡化和方便了原有的方法。現将最常用的應用領域及研究方法介紹如下。
基因分析
PCR技術能夠快速、靈敏地放大被測試的目的基因,所以可用于鑒定由于基因缺失、突變、轉位及外源基因進入(如病毒感染)所引起的各種疾病。PCR技術已廣泛地用于遺傳病的基因分析、産前診斷、傳染病病原體檢測、癌基因臨床分析等方面。PCR結合分子雜交等分析方法可進一步提高檢測的靈敏度和準确性。
定位克隆
隻要知道目的基因兩端的序列,就可通過RT-PCR和重組PCR等技術進行基因克隆,這不僅省略了通常制備DNA片段的繁瑣步驟,也避免了進行亞克隆的經典程序。
序列分析
PCR技術使DNA測序大為簡化,因此現在幾乎都采用PCR法進行序列測定。
除了上述三個方面的應用外,PCR還可用來制備高比活性标記探針,利用重組PCR可進行基因的人工定位突變和基因表達調控的研究。利用錨定PCR、反向PCR等還可對求知序列的基因進行分析。利用差示PCR、競争PCR可進行DNA及mRNA的定量分析。法醫學中應用PCR還可精确地選擇出組織器官移植的合适配型。