简介
1976年Segawa等首次描述该病,国外已有不少报道,近几年已引起中国临床工作者的高度重视。
反应性肌张力不全的异常基因位于第14号常染色体短臂,与四氢生物喋吟的产生有关。患者脑脊液中生物喋吟的含量降低。四氢生物喋吟是酪氨酸转化为多巴胺的重要辅助因子,其产生障碍使多巴胺合成受到影响。
病因
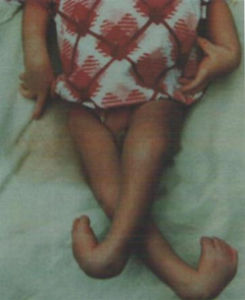
目前认为GTP环化水解酶的同工酶GCHⅠ缺乏导致多巴胺合成减少是DRD的主要病因(Nagatsu1998)。反应性肌张力不全常见于儿童期,女性多于男性。发病年龄一般在4-8岁,但可以早至婴儿期,晚至成人期。婴儿期起病者较少见,常被误诊为脑性瘫痪或痉挛性截瘫,成人期起病者症状类似帕金森病。初始症状往往是马蹄内翻足和由于下肢肌张力不全造成的步态异常。以后病情进行性加重,可以出现四肢僵硬,运动徐缓,面无表情。
半数病人出现8-10Hz位置性、意向性震颤(不同于帕金森病的4-5Hz静止性震颤),一般在成人期病情相对稳定。某些轻症病人仅在午后有行走困难和疲劳感,写字握笔略久有书写痉孪。体检时在某些患儿可发现踝阵挛和巴彬斯基征阳性。75%患儿的肌张力不全有特征性的昼间变化,即清晨刚起床时肌张力不全较轻,以后渐渐加重,黄昏时最为明显。日间休息后可稍有改善,但活动、运动后加剧。
临床表现
反应性肌张力不全常见于儿童期,女性多于男性。发病年龄一般在4~8岁,但可以早至婴儿期,晚至成人期。婴儿期起病者较少见,常被误诊为脑性瘫痪或痉挛性截瘫,成人期起病者症状类似帕金森病。初始症状往往是马蹄内翻足和由于下肢肌张力不全造成的步态异常。以后病情进行性加重,可以出现四肢僵硬,运动徐缓,面无表情。半数病人出现8-10Hz位置性、意向性震颤(不同于帕金森病的4-5Hz静止性震颤),一般在成人期病情相对稳定。某些轻症病人仅在午后有行走困难和疲劳感,写字握笔略久有书写痉挛。体检时在某些患儿可发现踝阵挛和巴彬斯基征阳性。75%患儿的肌张力不全有特征性的昼间变化,即清晨刚起床时肌张力不全较轻,以后渐渐加重,黄昏时最为明显。日间休息后可稍有改善,但活动、运动后加剧。
发病年龄
DRD发病年龄在1~12岁,占儿童肌张力障碍的10%,少数患者可在50~60岁发病。女性发病率多于男性,男女比例为1:4。缓慢起病,通常首发于下肢,表现为上肢或下肢的肌张力障碍和异常姿势或步态,步态表现为腿僵直、足屈曲或外翻,严重者可累及颈部。少数患者首发症状可为震颤。肌张力障碍也可合并运动迟缓、齿轮样肌强直、姿势反射障碍等帕金森综合征之表现。症状具有昼间波动,一般在早上或午后症状很轻微,运动后或晚间加重。此种现象随年龄增大会变得不明显,一般在起病后20年内病情进展明显,20~30年趋于缓和,至40年病情几乎稳定。
儿童起病者
多以一侧下肢肌张力异常为首发症状,患儿出现怪异步态下肢僵硬、步态不稳、马蹄内翻足等。有时仅表现学走路较迟易摔倒随病情发展,肌张力异常影响到其他肢体,甚至头颈部及身体中轴出现痉挛性斜颈扭转痉挛患儿可有肢体震颤、肌强直及自动Babinske征阳性,语言及智能一般不受累。
成人起病者
以肢体不自主震颤、僵硬感等类似帕金森综合征表现多见患者行动迟缓易疲劳,肢体肌张力增高腱反射亢进,病理征阳性。75%患者症状有昼夜波动性晨起或休息后明显减轻甚而消失下午或劳累后症状加重。
多数患者
多数病程呈进展性,未经治疗者最终生活不能自理。
并发症
流行性肌张力障碍的临床表现以短暂出现的口、眼、舌、颈等肌为主,可累及四肢躯干肌肉的肌张力障碍发作为特征,发作呈转颈、伸舌、两眼向一侧凝视或上翻以及四肢持续性伸展或屈曲姿势,也可出现快速抖动。通常每次发作数分至数小时,每日1~2次至十多次。发作后病人觉头昏、头痛、全身酸痛。
诊断依据
①有肌张力不全的临床表现,如肢体僵硬。步态异常等;
②症状轻重有明显的昼间变化,晨轻暮重,并在活动后加重;
③神经系统检查:肌力正常,部分患儿肌张力增高,踝阵挛阳性、巴彬斯基征阳性;
④小剂量左旋多巴治疗反应迅速,疗效持久。
诊断主要依据临床表现及对小剂量多巴制剂的反应性。儿童或成人起病,以原因不明的肢体肌张力异常、震颤步态怪异等为首发症状,晨轻暮重为主要临床特点,尤其有家族遗传史者应高度怀疑本病。可疑患者给予口服小剂量多巴制剂多数在1~3天症状缓解若无效,可适当增加剂量国外报道(Torbjoerna1991)卡比多巴/左旋多巴剂量增至25/100(含左旋多巴100mg及卡比多巴25mg),3次/d仍无效,即可排除DRD的诊断。
鉴别诊断
本病应与脑性瘫痪、少年型帕金森病、扭转痉挛、肝豆状核变性、痉挛性截瘫等鉴别。
1.脑性瘫痪常以肌张力异常增高及痉挛为主要特征但常伴智力低下、惊厥及情绪障碍症状无波动性对多巴制剂无反应。
2.少年型帕金森病极少发生在8岁以下儿童PET检查示18F-dopa摄取量下降,长期应用多巴制剂需逐渐增加剂量,且易出现异动、剂末恶化等副作用。
3.肝豆状核变性常伴肝脏损害及智力、精神异常,角膜可见K-F环。
4.极少数患者初始症状体征酷似痉挛性截瘫,小剂量多巴的戏剧性反应性可能是最重要的鉴别要点。
实验室检查
1.血尿便常规一般均正常。
2.脑脊液检查可正常,也有报道脑脊液高香草酸及生物喋呤含量降低。
3.肝功能检查正常有鉴别诊断意义。其它辅助检查:脑电图诱发电位、颅脑CT、MRI及PET检查等均正常。
治疗
左旋多巴/苄丝肼(美多巴)0.125~0.25g/d,分2~3次服用中国际左旋多巴/苄丝肼(美多巴)用量一般为62.5~187.5mg/d。有报道持续用药15年,未出现任何副作用(Harwood,1994)但若停药,症状即重新出现。
预防
有遗传背景者,预防显得更为重要。预防措施包括避免近亲结婚推行遗传咨询携带者基因检测及产前诊断和选择性人工流产等防止患儿出生。早期诊断早期治疗加强临床护理对改善患者的生活质量有重要意义。
目前本组患者有关DRD/HPD的辅助检查,包括脑脊液、EEG、各种诱发电位、头颅CT、MRI、正电子发射断层扫描(PET)均正常。部分患者有血清酶学增高,可能与肌肉强直收缩有关。因本病具有较明显的特征性,只要对本病有足够认识和重视,诊断应不难,但应注意与脑性瘫痪、少年型帕金森病、肝豆状核变性等鉴别。DRD预后良好,小剂量多巴制剂即有良好效果,国外所用剂量为左旋多巴1.0~1.5g/d、帕金宁控释片或美多巴0.125~0.250g/d,本组服用后两者左旋多巴平均剂量仅84.8mg/d。Markova等曾随访观察15~20年,无需增大剂量,长期应用无异动症、开关现象等副作用。对于因长期肌张力障碍所致的脊柱、足部畸形,可采用器械康复、功能锻炼等综合性方法帮助恢复。
流行病学
较罕见,据估计,日本和英国DRD的发病率为1/200万,但迄今为止,中国仅报道过1个家系,可能与对本病不熟悉有关,本组除1例外,误诊时间均较长,治疗前平均病程达12.3年。由于本病有特效治疗方法,如能早期诊断、早期治疗,患者可保持完全正常的生活质量。1988年美国明尼苏达地区调查显示广泛性肌张力障碍和局限性(身体某部位)肌张力障碍年发病率分别为0.2/10万和2.4/10万患病率分别为3.4/10万和30/10万东欧地区年发病率和患病率均为明尼苏达的2倍中国尚无肌张力障碍流行病学资料。